Pre-defined pipelines 
vsearch OTUs
This automated workflow is mostly based on vsearch (Rognes et. al 2016) [manual] to form OTUs and an OTU table. This input is the directory that contains per-sample fastq files (demultiplexed data).
Analyses step |
Default setting |
|---|---|
CUT PRIMERS (optional) |
– |
read_R1 = \.R1min_overlap = 12min_length = 32allow_merge_stagger = TRUEinclude only R1 = FALSEmax_diffs = 20max_Ns = 0max_len = 600keep_disjoined = FALSEfastq_qmax = 41 |
|
maxEE = 1maxN = 0minLen = 32max_length = undefinedqmax = 41qmin = 0maxee_rate = undefined |
|
pre_cluster = 0.98min_unique_size = 1denovo = TRUEreference_based = undefinedabundance_skew = 2min_h = 0.28 |
|
ITS Extractor (optional) |
organisms = allregions = allpartial = 50region_for_clustering = ITS2cluster_full_and_partial = TRUEe_value = 1e-2scores = 0domains = 2complement = TRUEonly_full = FALSEtruncate = TRUE |
OTU_type = centroidsimilarity_threshold = 0.97strands = bothremove_singletons = falsesimilarity_type = 2sequence_sorting = cluster_sizecentroid_type = similaritymax_hits = 1mask = dustdbmask = dust |
|
ASSIGN TAXONOMY with BLAST (optional) |
database_file = select a databasetask = blastnstrands = both |
DADA2 ASVs
This pre-defined workflow is based on the DADA2 tutorial to form ASVs and an ASV table. This input is the directory that contains per-sample fastq files (demultiplexed data).
primer removal step do not represent parts from the DADA2 tutorial. Nevertheless, it is advisable to remove primers before proceeding with ASV generation with DADA2.pipeline mode |
when do use |
|---|---|
FORWARD |
for paired-end Illumina data where amplicons
are expected to be in uniform orientation
(e.g., all reads are in 5’-3’ orientation).
|
MIXED |
for paired-end Illumina data where amplicons
are expected to be both, in 5’-3’ (forward)
and 3’-5’ (reverse) oriented.
In that mode,
CUT PRIMERS is mandatory,and generates separate directories for forward
and reverse oriented sequences, which will pass
DADA2 pipeline individually. After merging the paired ends,
the reverse oriented sequences are reverse complemented
and aggregated with the forward reads for chimera filtering
and ASV table generation. The output ASVs are all 5’-3’ oriented.
Here, make sure that the R1 and R2 identifiers are
\.R1 and \.R2 in the QUALITY FILTERING step
|
PACBIO |
for single-end PacBio data.
CUT PRIMERS step for single-enddata will reoriente all reads to 5’-3’ (forward) orientation.
|
Note
Working directory must contain at least 2 samples for DADA2 pipeline.
Default options:
Analyses step |
Default setting |
|---|---|
CUT PRIMERS (optional) |
–
|
read_R1 = \.R1read_R2 = \.R2maxEE = 2maxN = 0minLen = 20truncQ = 2truncLen = 0maxLen = 9999minQ = 2matchIDs = TRUE |
|
pool = FALSEselfConsist = FASLEqualityType = Auto |
|
minOverlap = 12maxMismatch = 0trimOverhang = FALSEjustConcatenate = FALSE |
|
method = consensus |
|
Filter ASV table (optional) |
collapseNoMismatch = TRUEby_length = 250minOverlap = 20vec = TRUE |
ASSIGN TAXONOMY (optional) |
minBoot = 50tryRC = FALSEdada2 database = select a database |
QUALITY FILTERING
DADA2 filterAndTrim function performs quality filtering on input FASTQ files based on user-selected criteria. Outputs include filtered FASTQ files located in the qualFiltered_out directory.
Quality profiles may be examined using the QualityCheck module.
Setting |
Tooltip |
|---|---|
|
applies only for paired-end data.
Identifyer string that is common for all R1 reads
(e.g. when all R1 files have ‘.R1’ string, then enter ‘\.R1’.
Note that backslash is only needed to escape dot regex; e.g.
when all R1 files have ‘_R1’ string, then enter ‘_R1’.).
|
|
applies only for paired-end data.
Identifyer string that is common for all R2 reads
(e.g. when all R2 files have ‘.R2’ string, then enter ‘\.R2’.
Note that backslash is only needed to escape dot regex; e.g.
when all R2 files have ‘_R1’ string, then enter ‘_R2’.).
|
|
discard sequences with more than the specified number of expected errors
|
|
discard sequences with more than the specified number of N’s (ambiguous bases)
|
|
remove reads with length less than minLen. minLen is enforced
after all other trimming and truncation
|
|
truncate reads at the first instance of a quality score less than or equal to truncQ
|
|
truncate reads after truncLen bases
(applies to R1 reads when working with paired-end data).
Reads shorter than this are discarded.
Explore quality profiles (with QualityCheck module) and
see whether poor quality ends needs to be truncated
|
|
applies only for paired-end data.
Truncate R2 reads after truncLen bases.
Reads shorter than this are discarded.
Explore quality profiles (with QualityCheck module) and
see whether poor quality ends needs to truncated
|
|
remove reads with length greater than maxLen.
maxLen is enforced on the raw reads.
In dada2, the default = Inf, but here set as 9999
|
|
after truncation, reads contain a quality score below minQ will be discarded
|
|
applies only for paired-end data.
If TRUE, then double-checking (with seqkit pair) that only paired reads
that share ids are outputted.
Note that ‘seqkit’ will be used for this process, because when
using e.g. SRA fastq files where original fastq headers have been
replaced, dada2 does not recognize those fastq id strings
|
see default settings
DENOISING
DADA2 dada function to remove sequencing errors.
Outputs filtered fasta files into denoised_assembled.dada2 directory.
Setting |
Tooltip |
|---|---|
|
if TRUE, the algorithm will pool together all samples prior to sample inference.
Pooling improves the detection of rare variants, but is computationally more expensive.
If pool = ‘pseudo’, the algorithm will perform pseudo-pooling between individually
processed samples.
|
|
if TRUE, the algorithm will alternate between sample inference and error rate estimation
until convergence
|
|
‘Auto’ means to attempt to auto-detect the fastq quality encoding.
This may fail for PacBio files with uniformly high quality scores,
in which case use ‘FastqQuality’
|
see default settings
MERGE PAIRS
DADA2 mergePairs function to merge paired-end reads.
Outputs merged fasta files into denoised_assembled.dada2 directory.
Setting |
Tooltip |
|---|---|
|
the minimum length of the overlap required for merging the forward and reverse reads
|
|
the maximum mismatches allowed in the overlap region
|
|
if TRUE, overhangs in the alignment between the forwards and reverse read are
trimmed off. Overhangs are when the reverse read extends past the start of
the forward read, and vice-versa, as can happen when reads are longer than the
amplicon and read into the other-direction primer region
|
|
if TRUE, the forward and reverse-complemented reverse read are concatenated
rather than merged, with a NNNNNNNNNN (10 Ns) spacer inserted between them
|
see default settings
CHIMERA FILTERING
DADA2 removeBimeraDenovo function to remove chimeras.
Outputs filtered fasta files into chimeraFiltered_out.dada2 and final ASVs to ASVs_out.dada2 directory.
Setting |
Tooltip |
|---|---|
|
‘consensus’ - the samples are independently checked for chimeras, and a consensus
decision on each sequence variant is made.
If ‘pooled’, the samples are all pooled together for chimera identification.
If ‘per-sample’, the samples are independently checked for chimeras
|
see default settings
filter ASV table
DADA2 collapseNoMismatch function to collapse identical ASVs;
and ASVs filtering based on minimum accepted sequence length (custom R functions).
Outputs filtered ASV table and fasta files into ASVs_out.dada2/filtered directory.
Setting |
Tooltip |
|---|---|
|
collapses ASVs that are identical up to shifts or
length variation, i.e. that have no mismatches or internal indels
|
|
discard ASVs from the ASV table that are shorter than specified
value (in base pairs). Value 0 means OFF, no filtering by length
|
|
collapseNoMismatch setting. Default = 20. The minimum overlap of
base pairs between ASV sequences required to collapse them together
|
|
collapseNoMismatch setting. Default = TRUE. Use the vectorized
aligner. Should be turned off if sequences exceed 2kb in length
|
see default settings
ASSIGN TAXONOMY
DADA2 assignTaxonomy function to classify ASVs.
Outputs classified fasta files into taxonomy_out.dada2 directory.
Setting |
Tooltip |
|---|---|
|
the minimum bootstrap confidence for assigning a taxonomic level
|
|
the reverse-complement of each sequences will be used for classification
if it is a better match to the reference sequences than the forward sequence
|
|
select a reference database fasta file for taxonomy annotation
|
see default settings
UNOISE ASVs
UNOISE3 pipeline for making ASVs (zOTUs). Can optionally do automatic clustering of those ASVs (zOTUs) to OTUs by specifying the similarity threshold < 1. Uses UNOISE3 and clustering algorithms in vsearch.
NextITS
NextITS is an automated pipeline for analysing full-length ITS reads obtained via PacBio sequencing.
Please see other details here: https://next-its.github.io Please note that NextITS pipeline accepts only a single primer pair, i.e., one forward and one reverse primer in STEP_1!
Important
NextITS requires your data and folders to be structured in a specific way (see below)!
Directory my_dir_for_NextITS contains Input [hard-coded requirement here] and one or multiple sequencing runs.
In the below example, the sequencing runs [RunID] are named as Run1, Run2 and Run3 (but naming can be different).
In PipeCraft2, following the examples below, select my_dir_for_NextITS as a WORKDIR.
Single sequencing run
my_dir_for_NextITS as a WORKDIR in PipeCraft2.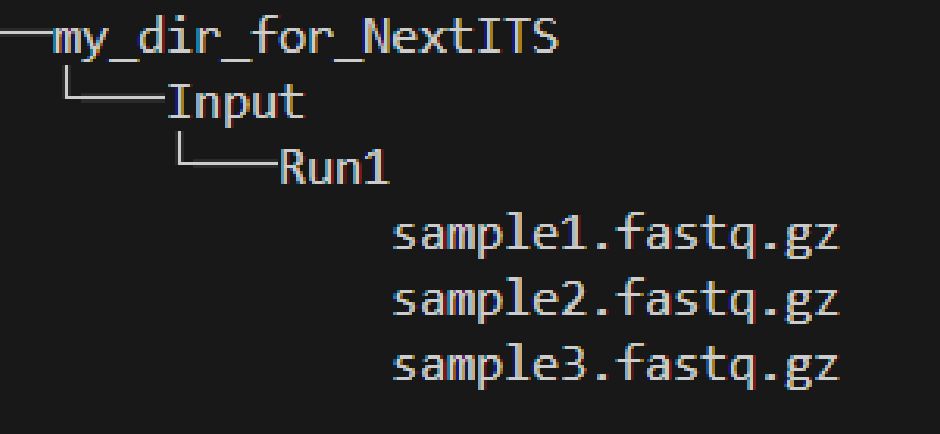
Input data for this pipeline must be demultiplexed, if your data is multiplexed use the demultiplexer from QuickTools before running the pipeline.
Sample naming
Please avoid non-ASCII symbols in SampleID,
and do not use the period symbol (.), as it represents the wildcard character in regular expressions.
Also, it is preferable not to start the sample name with a number.
Multiple sequencing runs
my_dir_for_NextITS as a WORKDIR in PipeCraft2.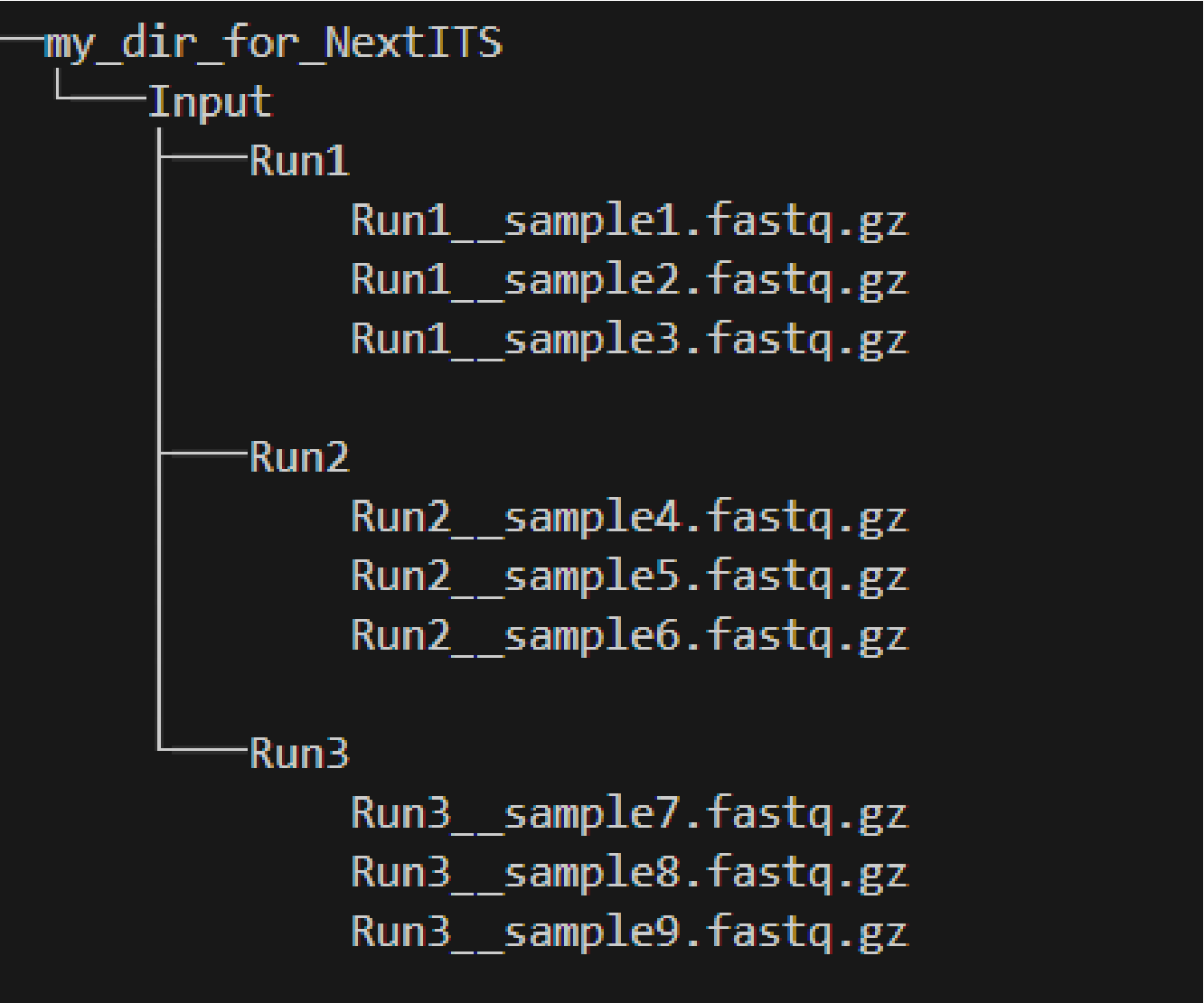
Input data for this pipeline must be demultiplexed, if your data is multiplexed use the demultiplexer from QuickTools before running the pipeline.
Sample naming
Please avoid non-ASCII symbols in RunID and SampleID,
and do not use the period symbol (.), as it represents the wildcard character in regular expressions.
Also, it is preferable not to start the sample name with a number.
NextITS uses the SequencingRunID__SampleID naming convention (please note the double underscore separating RunID and SampleID parts).
This naming scheme allows to easily trace back sequences, especially if the same sample was sequenced several times and is present in multiple sequencing runs.
In the later steps, extracting the SampleID part and summarizing read counts for such samples is easy.
Default settings:
Analyses step |
Default setting |
|---|---|
primer_mismatch = 2its_region = fullqc_maxhomopolymerlen = 25qc_maxn = 4ITSx_evalue = 1e-2ITSx_partial = 0ITSx_tax = allchimera_rescue_occurrence = 2tj f = 0.01tj p = 1hp = TRUE |
|
otu_id = 0.98swarm_d = 1lulu = TRUEunoise = FALSEotu_id_def = 2otu_qmask = dustswarm_fastidious = TRUEunoise_alpha = 2unoise_minsize = 8max_MEEP = 0.5max_chimera_score = 0.5lulu_match = 95lulu_ratio = 1lulu_ratiotype = minlulu_relcooc = 0.95lulu_maxhits = 0 |
{kind=link}
{kind=link}
Cut primers
Please note that NextITS pipeline accepts only a single primer pair, i.e., one forward and one reverse primer!
Setting |
Tooltip |
|---|---|
|
Specify forward primer, IUPAC codes allowed
|
|
Specify reverse primer, IUPAC codes allowed
|
|
Specify allowed number of mismatches for primers
|
Quality filtering
Filter sequences based on expected errors per sequence and per base, compress and correct homopolymers.
Setting |
Tooltip |
|---|---|
|
Maximum number of expected errors
|
|
Maximum number of expected error per base
|
|
Discard sequences with more than the specified number of ambiguous nucleotides (N’s)
|
|
Threshold for a homopolymer region lenght in a sequence
|
|
Enable or disable homopolymer correction
|
ITS extraction
these conserved regions don’t offer species-level differentiation.
random errors in these areas can disrupt sequence clustering.
chimeric breakpoints, which are common in these regions, are hard to detect in short fragments ranging from 10 to 70 bases.
NextITS deploys the ITSx software (Bengtsson-Palme et al. 2013) for extracting the ITS sequence.
Setting |
Tooltip |
|---|---|
|
ITS part selector (ITS1, ITS2 or full)
|
|
Taxonomy profile for ITSx can be used to restrict the search to only taxon(s) of interest.
|
|
E-value cutoff threshold for ITSx
|
|
Keep partial ITS sequences (specify a minimum length cutoff)
|
Chimera filtering
Additional step in NextITS is a “rescue” of sequences that were initially flagged as chimeric, but are occur at least in 2 samples (which represent independent PCR reactions); thus are likely false-positive chimeric sequences. The chimeric sequence occurrence frequency can be edited using the –chimera_rescue_occurrence parameter.
Setting |
Tooltip |
|---|---|
|
Database for reference based chimera removal (UDB)
|
|
A minimum occurence of initially flagged chimeric sequence required to rescue them
|
Tag-jump correction
Tag-jumps, sometimes referred to as index-switches or index cross-talk, may represent a significant concern in high-throughput sequencing (HTS) data. They can cause technical cross-contamination between samples, potentially distorting estimates of community composition. Here, tag-jump events are evaluated the UNCROSS2 algorithm (Edgar 2018 ) are removed.
Setting |
Tooltip |
|---|---|
|
UNCROSS parameter f for tag-jump filtering
|
|
UNCROSS parameter p for tag-jump filtering
|
UNOISE denoising
The UNOISE algorithm (Edgar 2016 ) focuses on error-correction (or denoising) of amplicon reads. Essentially, UNOISE operates on the principle that if a sequence with low abundance closely resembles another sequence with high abundance, the former is probably an error. This helps differentiate between true biological variation and sequencing errors. It’s important to note that UNOISE was initially designed and optimized for Illumina data. Because of indel errors stemming from inaccuracies in homopolymeric regions, UNOISE might not work well with data that hasn’t undergone homopolymer correction.
Setting |
Tooltip |
|---|---|
|
Enable or disable denoising with UNOISE algorithm
|
|
Alpha parameter for UNOISE
|
|
Minimum sequence abundance
|
Clustering
NextITS supports 3 different clustering methods:
vsearch: this employs greedy clustering using a fixed sequence similarity threshold with VSEARCH (Rognes et al., 2016, );
swarm: dynamic sequence similarity threshold for clustering with SWARM (Mahé et al., 2021, );
unoise: creates zero-radius OTUs (zOTUs) based on the UNOISE3 algorithm (Edgar 2016 );
Setting |
Tooltip |
|---|---|
|
Sequence clustering method (choose from: vsearch, swarm, unoise)
|
|
Sequence similarity threshold
|
|
Sequence similarity definition (applied to UNOISE as well)
|
|
Method to mask low-complexity sequences (applied to UNOISE as well)
|
|
SWARM clustering resolution (d)
|
|
Link nearby low-abundance swarms (fastidious option)
|
Post-clustering with LULU
The purpose of LULU is to reduce the number of erroneous OTUs in OTU tables to achieve more realistic biodiversity metrics. By evaluating the co-occurence patterns of OTUs among samples LULU identifies OTUs that consistently satisfy some user selected criteria for being errors of more abundant OTUs and merges these OTUs.
Setting |
Tooltip |
|---|---|
|
Enable or disable post-clustering curation with lulu
|
|
Minimum similarity threshold
|
|
Minimum abundance ratio
|
|
Abundance ratio type - “min” or “avg
|
|
Relative co-occurrence
|
|
Maximum number of hits (0 = unlimited)
|