DADA2 ASVs pipeline, COI 
This example dataset consists of COI mtDNA gene amplicon sequences with the target length of 313 bp:
For this example data run, we are using a subset of CO1Classifier database in the taxonomy annotation process, download it from here.
Starting point
This example dataset consists of COI mtDNA gene amplicon sequences with the target length of 313 bp:
paired-end Illumina MiSeq data;
demultiplexed set (per-sample fastq files);
primers are not not removed;
sequences in this set are 5’-3’ (fwd) oriented.
when working with your own data …
… then please check that the paired-end data file names contain R1 and R2 strings (not just _1 and _2), so that PipeCraft can correctly identify the paired-end reads.
At least 2 samples (2x R1 + 2x R2 files) are required for this workflow! Otherwise ERROR in the denoising step:

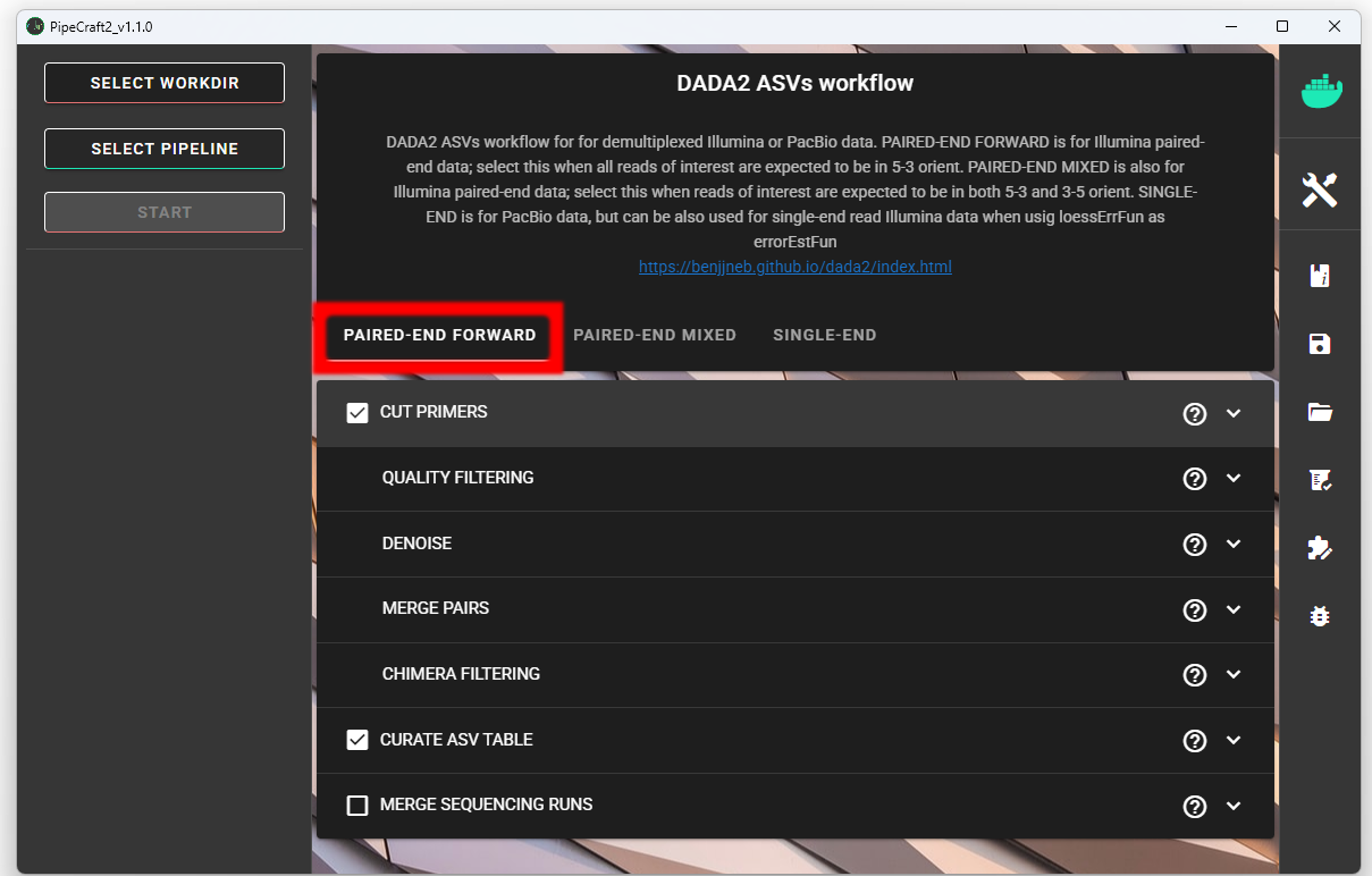
SELECT PIPELINE –> DADA" ASVs.
SELECT WORKDIRsequence files extension as *.fastq;sequencing read types as paired-end.Workflow mode
Because we are working with sequences that are 5’-3’ oriented, we are selecting hte PAIRED-END FORWARD mode of the pipeline.
if sequences are in mixed orientation
If some sequences in your library are in 5’-3’ and some as 3’-5’ orientation, then with the ‘PAIRED-END FORWARD’ mode exactly the same ASV may be reported twice, where one ASV is just the reverse complementary of another. To avoid that, select PAIRED-END MIXED mode. Sequences have mixed orientation in libraries where sequenceing adapters have been ligated, rather than attached to amplicons during PCR.
Specifying primers (for CUT PRIMERS) is mandatory for the PAIRED-END MIXED mode. Based on the priemr sequences, the library will be split into two: 1) fwd oriented sequences, and 2) rev oriented sequences. Both batches are processed independently to produce ASVs, after which the rev oriented batch ASVs are reverse complemented and merged with the fwd oriented ASVs. Identical ASVs are merged to form a final data set. This is a reccomended workflow for accurate denoising compared with first reorienting all sequences to 5’-3’, and then performing a standard ‘PAIRED-END FORWARD’ workflow.
Cut primers
The example dataset contains primer sequences. Generally, we need to remove these to proceed the analyses only with the variable metabarcode of interest. If there are some additional sequence fragments, from eg. sequencing adapters or poly-G tails, then clipping the primers will remove those fragments as well.
Tick the box for CUT PRIMERS and specify forward and reverse primers.
For the example data, the forward primer is GGWACWGGWTGAACWGTWTAYCCYCC and reverse primer is TANACYTCNGGRTGNCCRAARAAYCA.
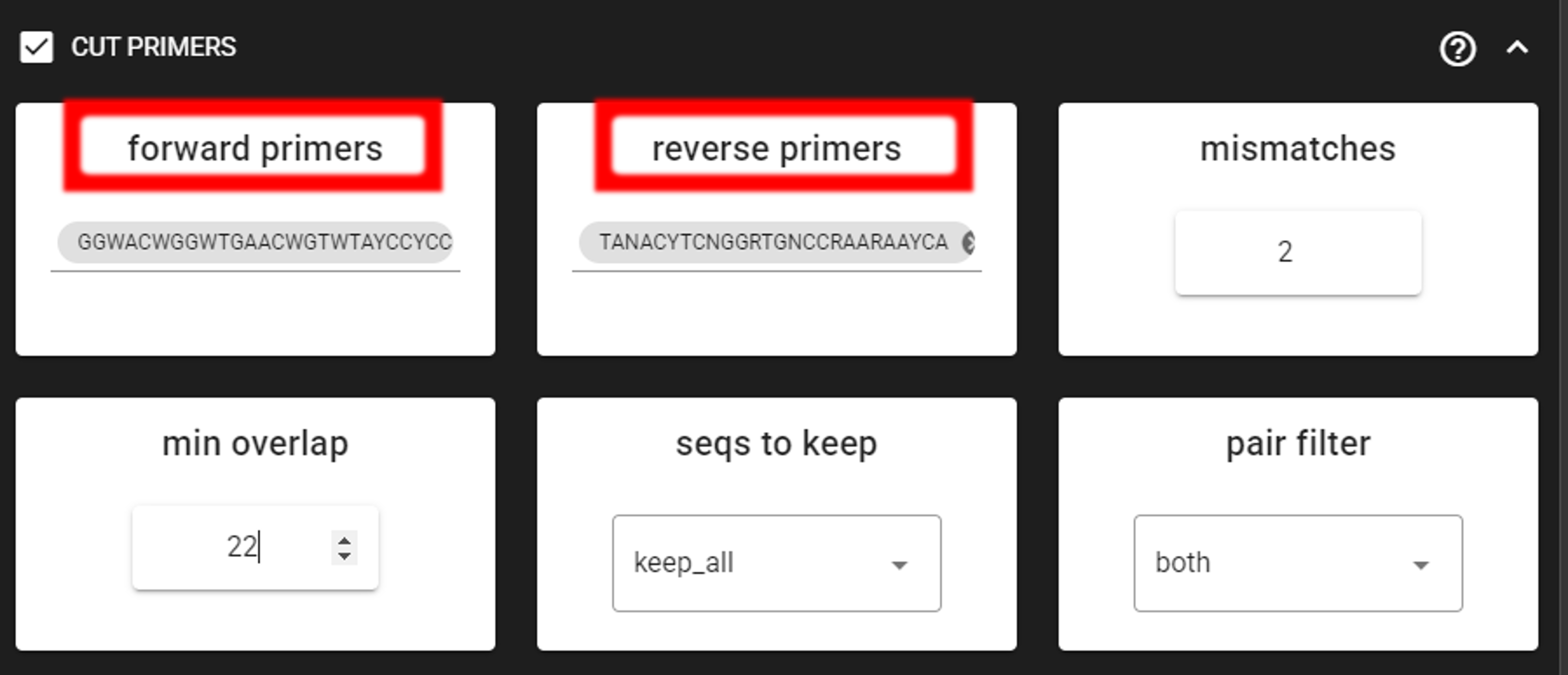
The primers are 26 bp - to keep a bit of flexibility in the primer search, we are requesting the min overlap of 22 bp and are allowing maximum of 2 mismatches .
Note that too low min overlap may lead to random matches. Check other CUT PRIMER options here.
Quality filtering
Before adjusting quality filtering settings, let’s have a look on the quality profile of our example data set.
Below quality profile plot was generated using QualityCheck panel (see here).
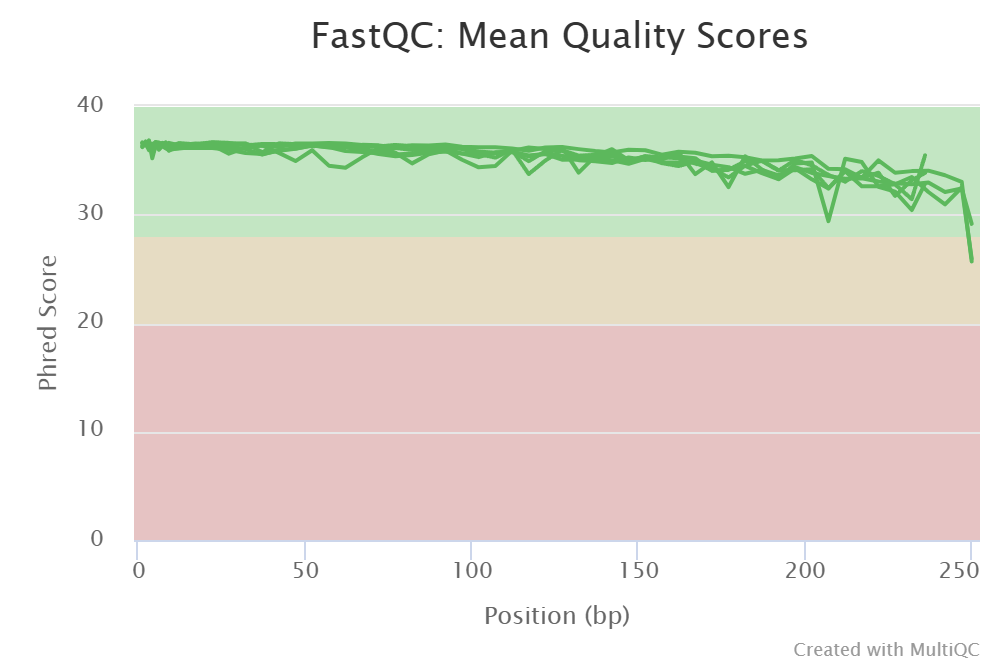
All files are represented with green lines, indicating good average quality per file (i.e., sample).
However, if you see lower qualities of especially towards the end of R2 reads, then it not too alarming, since
those can be clipped off with truncLen R2 setting (see remove low-quality ends/starts of reads section).
DADA2 algoritm is robust to lower quality sequences,
but removing the low quality read parts will improve the DADA2 sensitivity to rare sequence variants.
But herein, we do not need to clip the ends, because the overall quality of the sequences is good enough.
Click on QUALITY FILTERING to expand the panel
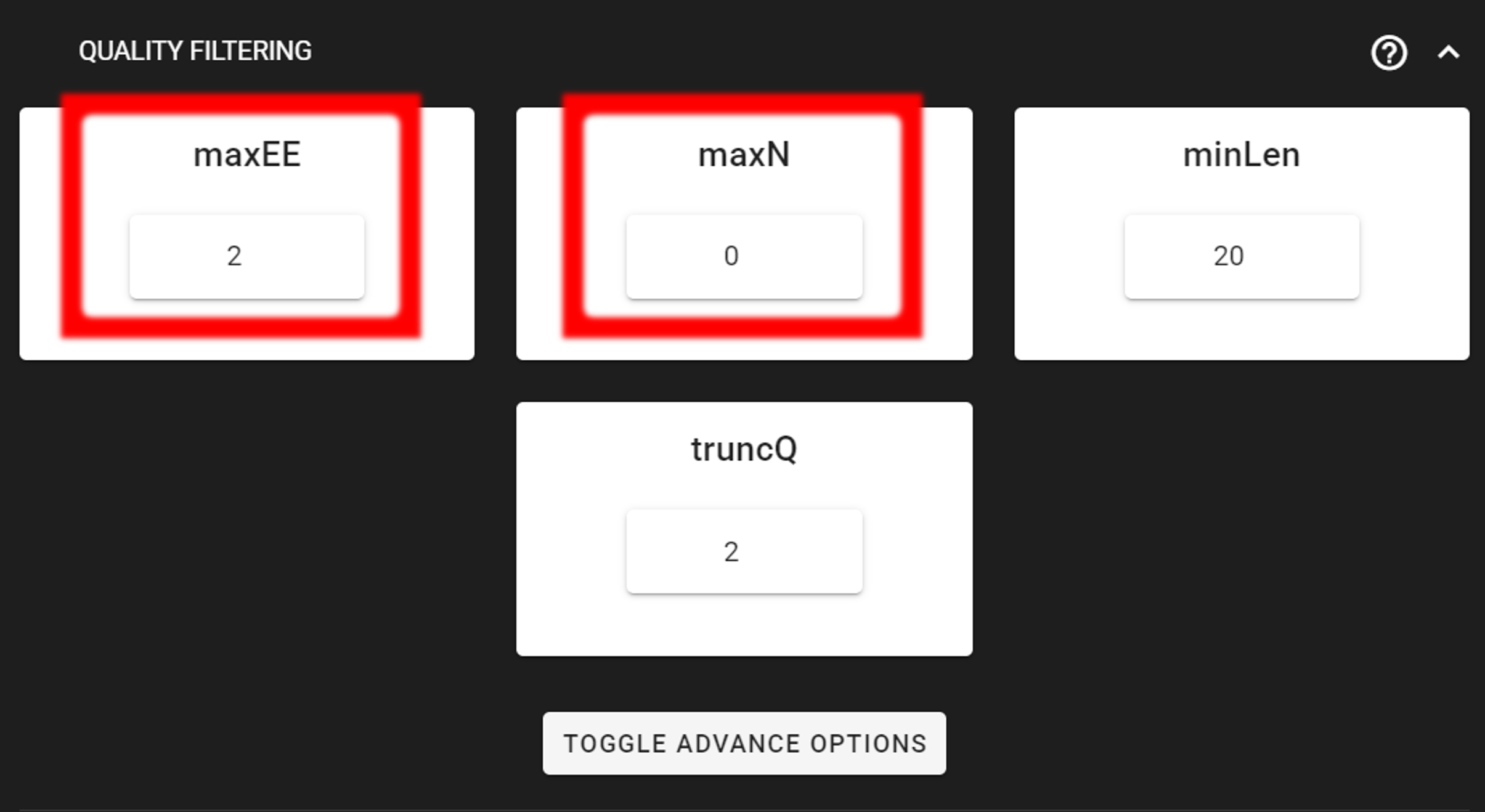
Here, we can leave the settings as DEFAULT by discarding sequences with maximum error rate of >2 and with ambiguous bases of >0.
Output directory |
|
|---|---|
*.fq.gz |
quality filtered sequences per sample in FASTQ format |
*.rds |
R objects for the following DADA2 workflow processes |
seq_count_summary.csv |
summary of sequence counts per sample |
Denoise and merge pairs
This step performs desiosing (as implemented in DADA2), which first forms ASVs per R1 and R2 files. Then during merging/assembling process the paired ASV mates are assembled to output full amplicon length ASV.

Here, we are working with Illumina data, so let’s make sure that the errorEstFun setting is loessErrfun.
We can leave all settings as DEFAULT.
Output directory |
|
|---|---|
*.fasta |
denoised and assembled ASVs per sample in FASTA format |
*.rds |
R objects for the following DADA2 workflow processes |
Error_rates_R*.pdf |
plots for estimated R1/R2 error rates |
seq_count_summary.csv |
summary of sequence counts per sample |
Chimera filtering
This step performs chimera filtering according to DADA2 removeBimeraDenovo function. During this step, the ASV table is also generated.
Important
make sure that primers have been removed from your amplicons; otherwise many false-positive chimeras may be filtered out from your dataset.
Here, we filter chimeras using the consensus method.
Output directory |
|
|---|---|
*.fasta |
chimera filtered ASVs per sample |
seq_count_summary.csv |
summary of sequence counts per sample |
‘chimeras’ dir |
ASVs per sample identified as chimeras |
Output directory |
|
|---|---|
ASVs_table.txt |
denoised and chimera filtered ASV-by-sample table |
ASVs.fasta |
corresponding FASTA formated ASV Sequences |
ASVs per sample identified as chimeras |
rds formatted denoised and chimera filtered ASV table |
Curate ASV table
This process first removes putative tag jumps and then collapses the ASVs that are identical up to shifts or length variation, i.e. ASVs that have no internal mismatches (PipeCraft2 uses vsearch usearch_global –id 1 for that); and finally filters out ASVs that are shorter/longer than specified length (in base pairs).
Here, we are enabling this process by checking the box for CURATE ASV TABLE in the DADA2 ASV workflow panel.
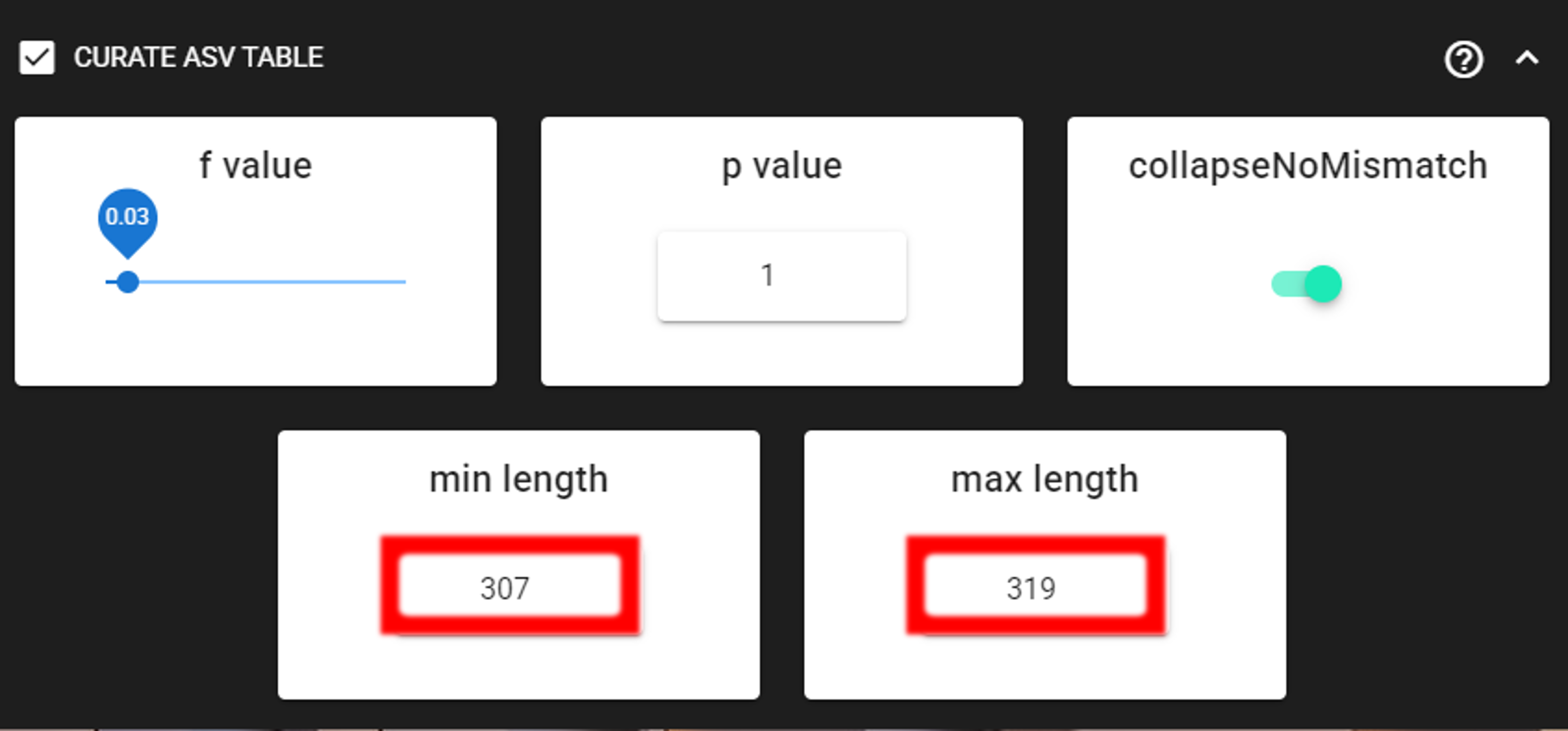
The f_value and p_value settings are used to filter out putative tag jumps (using UNCROSS2 algorithm).
Generally, we recommend to use p_value of 1 (default), and f_value of 0.03 when using combinational indexing strategy;
f_value of 0.05 when using single-indexes, and f_value of 0.01 when using unique dual-indexes.
The expected amplicon length (without primers) in our example dataset in 313 bp.
Assuming that shorter sequences are non-target sequences,
we use 307 in the min length setting and 319 in max length setting.
This will discard ASVs that are shorter than 307 bp or longer than 319 bp.
We are also setting the collapseNoMismatch to TRUE, to collapse identical ASVs.
This is basically equivalent to 100% clustering by ignoring the end gaps.
Output directory |
|
|---|---|
ASVs_table_TagJumpFilt.txt |
only tag-jump filtered ASV-by-sample table |
ASVs.fasta |
corresponding ASV Sequences with ASVs_table_TagJumpFilt.txt table |
ASVs_collapsed.fasta
|
tag-jump filtered and collapsed and size filtered
ASV Sequences. Present only if some ASVs were collapsed.
|
ASVs_table_collapsed.txt
|
corresponding ASV-by-sample table.
Present only if some ASVs were collapsed.
|
TagJump_stats.txt |
summary of tag-jump filtering results |
If there is nothing to collapse or filter out based on the length
then there are no corresponding files in the ASVs_out.dada2/curated directory, and only
ASVs_table_TagJumpFilt.txt and ASVs.fasta files will be generated
(even when there is nothing to tag-jump filter - in which case ASVs_table_TagJumpFilt.txt is the same
ASVs_table.txt in the ASVs_out.dada2 directory).
Note
The pre-compiled pipeline ends here. Outputs COI ASVs should be further filtered to remove putative pseudogenes (NUMTs), and optionally ASVs can be clustered into OTUs. See below.
Save workflow
Once we have decided about the settings in our workflow, we can save the configuration file by pressing save workflow button on the right-ribbon

If you forget the save, then no worries, a pipecraft2_last_run_configuration.json file will be generated
for you upon starting the workflow.
As the file name says, it is the workflow configuration file for your last PipeCraft run in this working directory.
If the file name (pipecraft2_last_run_configuration.json) is not changed, then the file is overwritten with the new configuration
if running a new job in the same working directory.
This JSON file can be loaded into PipeCraft2 to automatically configure your next runs exactly the same way.
Note
‘Assign taxonomy’ is not the part of the full per-defined pipeline. This step can be selected and run via QuickTools panel. See below.
Start the workflow
Press START on the left ribbon to start the analyses.
when running the module for the first time …
… a docker image will be first pulled to start the process.
For example: 
When you need to STOP the workflow, press STOP button 
When the workflow has completed …
… a message window will be displayed.

Assign taxonomy
Assign taxonomy is not the part of the full per-defined pipeline, but can be run as a separate step in QuickTools. Here, we are using the SINTAX classifier for that.
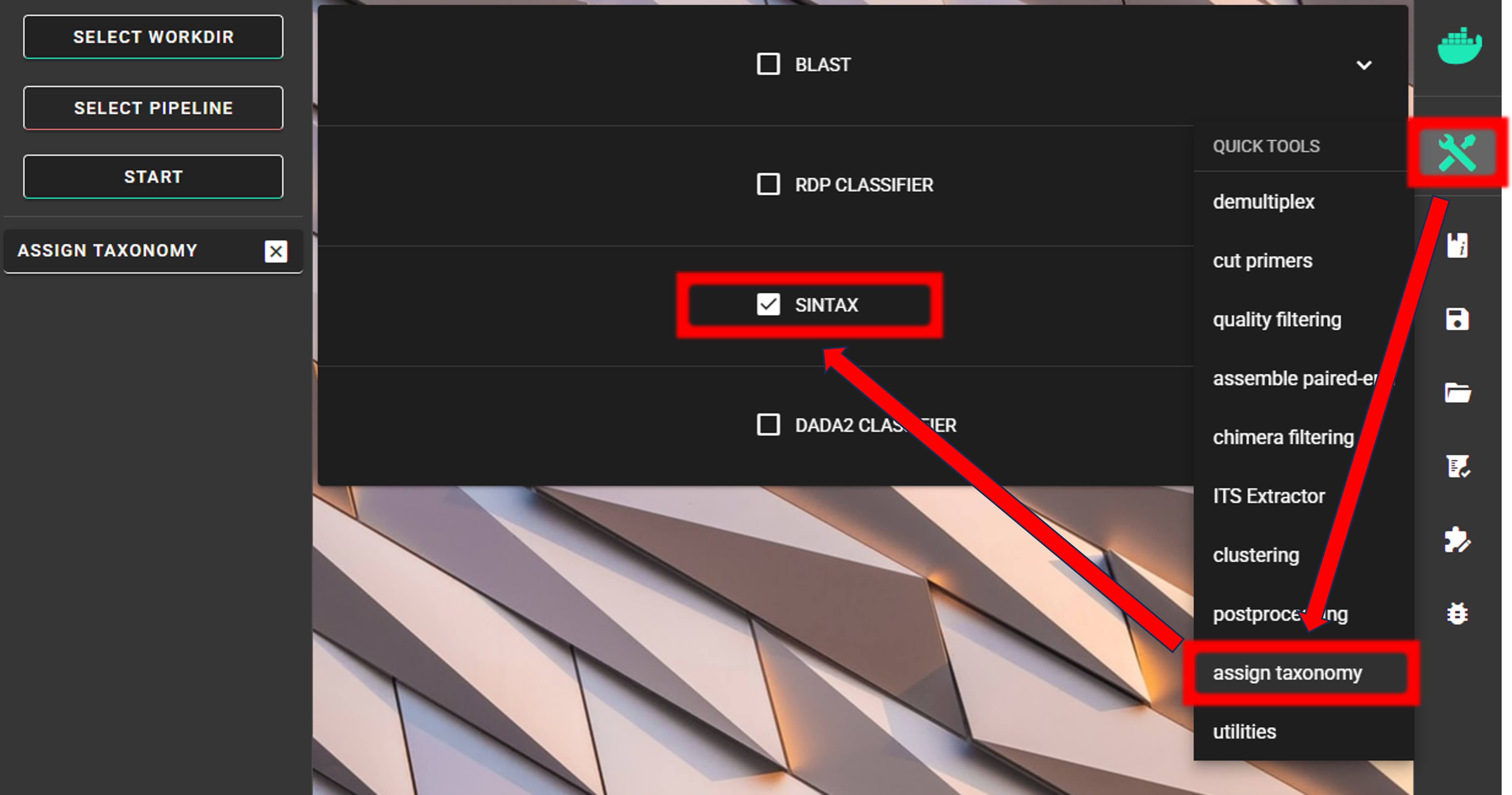
We need to specify the location of the reference DATABASE for the taxonomic classification of our ASVs. For this example data run, we are using a subset of CO1Classifier database in the taxonomy annotation process, download it from here
Specify the location of your downloaded database and also the fasta file with ASVs (fasta file) to be classified.
Herein, we use ASVs_collapsed.fasta file in the ASVs_out.dada2/curated directory
(since we applied also CURATE ASV TABLE process (see below Examine the outputs section)).
We can use the default cutoff (minimum bootstrap; ranging from 0-1; ~assignment confidence) value of 0.8.
This means that taxonomic ranks with at least bootstrap value of 80 will get classification (unclassified for <0.8).
strand may be plus (since we are expecting only 5’-3’ oriented ASVs),
but since SINTAX is fast, I’ll leave it as default (both - comparre both strands).
To START
To START, specify working directory under SELECT WORKDIR (outputs will be written here),
but the following requests about Sequence files extension and Sequencing read types do not matter here, just click ‘Confirm’.
Note
First time usage of the fasta formatted database requires conversion to the SINTAX database format (.udb). This conversion is performed automatically by PipeCraft2, and will take some time, depending on the size of the database.
Output directory |
|
|---|---|
taxonomy.sintax.txt |
classifier results with bootstrap values |
Examine the outputs
Several process-specific output folders are generated ![]()
|
paired-end fastq files per sample, primers clipped |
|
quality filtered paired-end fastq files per sample |
|
denoised and assembled fasta files per sample |
|
chimera filtered fasta files per sample |
|
ASVs table, and ASV sequences files |
|
curated ASVs table, and ASV sequences files |
|
ASVs taxonomy table (taxonomy.csv) |
Each folder (except ASVs_out.dada2 and taxonomy_out.dada2) contain
summary of the sequence counts (seq_count_summary.csv).
Examine those to track the read counts throughout the pipeline.
For example, from the seq_count_summary.csv file in qualFiltered_out
we see that most of the sequences survived the quality filtering step.
The input column represents the number of input sequences for the quality filtering step (that is, sequences from CUT PRIMERS step);
the qualFiltered column represents the number of sequences that survived the quality filtering step.
input |
qualFiltered |
|
sample1_COI |
999 |
830 |
sample2_COI |
999 |
829 |
sample3_COI |
999 |
871 |
Final outputs of the pipeline
Here, we applied also “CURATE ASV TABLE” process.
Therefore, our final outputs of the pipeline are in the ASVs_out.dada2/curated directory, which contans:
ASVs_table_TagJumpFilt.txt |
only tag-jump filtered ASV-by-sample table |
ASVs.fasta |
corresponding ASV Sequences with ASVs_table_TagJumpFilt.txt table |
ASVs_collapsed.fasta
|
tag-jump filtered and collapsed and size filtered
ASV Sequences.
|
ASVs_table_collapsed.txt |
corresponding ASV-by-sample table. |
TagJump_stats.txt |
summary of tag-jump filtering results |
Important
COI amplicons should be also checked for the presence of putative pseudogenes (NUMTs). (see below Remove NUMTs section)).
If we see the ASVs_table_collapsed.txt in the ASVs_out.dada2/curated directory,
then this means that some ASVs were collapsed and/or discarded because of the length filtering.
Let’s check the README.txt file:
there we can read that input ASV table (ASVs_out.dada2/ASVs_table.txt) had 27 ASVs and the output
ASVs table (ASVs_out.dada2/curated/ASVs_table_collapsed.txt) had 23 ASVs. Further, it says that
lenFilt resulted in 23 Features (ASVs).
This means that 4 ASVs were discarded because of the length filtering not because some were collapsed.
The length filtering and collapsing has been performed upon tag-jump filted ASV table (ASVs_table_TagJumpFilt.txt),
therefore, the final outputs of the pipeline are ASVs_table_collapsed.txt and ASVs_collapsed.fasta
ASVs_table_collapsed.txt represents the ASV table after the tag-jump and lenght/collapse filtering,
where the 1st column represents ASV identifiers (sha1 encoded),
2nd column is the sequence of an ASV,
and all the following columns represent number of sequences in the corresponding samples
(sample name is taken from the file name). This is tab delimited text file.
ASVs_table_collapsed.txt:
OTU |
Sequence |
sample1_COI |
sample2_COI |
sample3_COI |
e837216e5… |
TTTATCTT… |
0 |
0 |
682 |
46a9fb279… |
ACTATCCTC… |
583 |
0 |
0 |
37838deee… |
TTAGCAGGG.. |
0 |
342 |
0 |
c787bfb1f… |
TCTTGCAA… |
215 |
0 |
0 |
Note: even though the ASVs column header is “OTU”, it represents ASVs as we preformed an ASVs workflow!
We applied also tag-jump filtering process (via f_value and p_value settings).
When checking the TagJump_stats.txt file in the ASVs_out.dada2/curated directory,
we see that based on our settings, 0 tag-jump events were detected.
[Therefore, ASVs_table_TagJumpFilt.txt and ASVs_out.dada2/ASVs_table.txt files are the same.]
Result from the taxonomy annotation process - taxonomy table (taxonomy.sintax.txt) - is located at the
taxonomy_out.sintax directory.
Taxonomy results for the first 4 ASVs
24a03fcd59d40823dcc5aacb594e9fc6b68bcf6b |
d:Eukaryota(1.00),k:Metazoa(1.00),p:Arthropoda(0.97),c:Insecta(0.31),o:Coleoptera(0.26),f:Curculionidae(0.17),g:Laparocerus(0.17),s:Laparocerus_exiguus(0.09) |
|
d:Eukaryota,k:Metazoa,p:Arthropoda |
46a9fb279afe3d45b304409d09d63e9181f94096 |
d:Eukaryota(1.00),k:Metazoa(1.00),p:Arthropoda(1.00),c:Chilopoda(1.00),o:Lithobiomorpha(1.00),f:Lithobiidae(1.00),g:Lithobius(1.00),s:Lithobius_curtipes(1.00) |
|
d:Eukaryota,k:Metazoa,p:Arthropoda,c:Chilopoda,o:Lithobiomorpha,f:Lithobiidae,g:Lithobius,s:Lithobius_curtipes |
c787bfb1fb95d92352fe1579dcc1a8f67c0d5ebe |
d:Eukaryota(1.00),k:Metazoa(1.00),p:Annelida(1.00),c:Clitellata(1.00),o:Enchytraeida(1.00),f:Enchytraeidae(1.00),g:Fridericia_segmented_worms(1.00),s:Fridericia_eiseni(0.99) |
|
d:Eukaryota,k:Metazoa,p:Annelida,c:Clitellata,o:Enchytraeida,f:Enchytraeidae,g:Fridericia_segmented_worms,s:Fridericia_eiseni |
e837216e5c192a25cf3808ba39f29c34b3fe00e9 |
d:Eukaryota(1.00),k:Metazoa(0.93),p:Arthropoda(0.92),c:Insecta(0.52),o:Lepidoptera(0.26),f:Geometridae(0.09),g:Homospora(0.06),s:Homospora_rhodoscopa(0.06) |
|
d:Eukaryota,k:Metazoa,p:Arthropoda |
taxonomy.sintax.txt is a tab-delimited text file without the initial header row:
1st column: ASV identifier (sha1 encoded)
2nd column: SINTAX classification result with bootstrap values in parentheses
3rd column: “+” sign
4th column: SINTAX classification result when considering the
cutoffvalue (minimum bootstrap of 0.8)
Check for the non-target hits
It is often the case that universal metabarcoding primers amplify also non-target DNA regions and/or non-target taxa. Working with this example dataset, we are interesed only in Metazoa (Animals), thus we should get rid of the off-target noise before proceeding with relevant statistical analyses.
When we carefully examine the results, the taxonomy table, then we can see that 1 ASV is classifed to Fungi, 1 ASVs is unclassified to kingdom level (kingdom le). But even among the Metazoa, there are some off-target hits; 1 ASV is classified as Chordata (specifically as Homo sapiens, human). We are not interested in latter as well. We should remove all of those off-target ASVs.
Below, you can find a R script to clean and organize sintax taxonomy table, as well as clean ASV table and ASVs fasta file from the off-target taxa.
1#!/usr/bin/env Rscript
2### Filter dataset based on SINTAX results to include target taxa
3
4# specify taxon and threshold
5target="Metazoa" # target taxonomic group(s);
6 # multiple groups should be from the same taxonomic level
7 # separator is "," (e.g., "Hymenoptera, Lepidoptera")
8tax_level="kingdom" # allowed levels: kingdom | phylum | class | order | family | genus
9off_targets = c("Chordata") # a list of off-targets within target
10threshold="0.8" # threshold for considering an ASV as a target taxon
11species_threshold = 0.9 # threshold for species level identification
12
13# specify the ASV table and ASVs.fasta file that would be filtered to include only target taxa
14ASV_fasta = "ASVs_collapsed.fasta"
15ASV_table = "ASVs_table_collapsed.txt"
16
17# specify the SINTAX-classifier output file (taxonomy file)
18taxtab="taxonomy.sintax.txt"
19
20#--------------------------------------#
21library(stringr)
22library(dplyr)
23library(Biostrings)
24
25# Function to parse SINTAX taxonomy format from vsearch output
26parse_sintax = function(tax_string) {
27 # Initialize result with NAs
28 result = list(
29 kingdom = NA, kingdom_conf = 0,
30 phylum = NA, phylum_conf = 0,
31 class = NA, class_conf = 0,
32 order = NA, order_conf = 0,
33 family = NA, family_conf = 0,
34 genus = NA, genus_conf = 0,
35 species = NA, species_conf = 0
36 )
37
38 if (is.na(tax_string) || tax_string == "" || tax_string == "*") {
39 return(result)
40 }
41
42 # Split by comma
43 ranks = strsplit(tax_string, ",")[[1]]
44
45 for (rank in ranks) {
46 # Extract rank prefix (d:, k:, p:, c:, o:, f:, g:, s:)
47 if (grepl("^d:", rank)) {
48 # Domain (skip, not used)
49 next
50 } else if (grepl("^k:", rank)) {
51 # Kingdom
52 match = regmatches(rank, regexec("k:([^(]+)\\(([0-9.]+)\\)", rank))[[1]]
53 if (length(match) == 3) {
54 result$kingdom = match[2]
55 result$kingdom_conf = as.numeric(match[3])
56 }
57 } else if (grepl("^p:", rank)) {
58 # Phylum
59 match = regmatches(rank, regexec("p:([^(]+)\\(([0-9.]+)\\)", rank))[[1]]
60 if (length(match) == 3) {
61 result$phylum = match[2]
62 result$phylum_conf = as.numeric(match[3])
63 }
64 } else if (grepl("^c:", rank)) {
65 # Class
66 match = regmatches(rank, regexec("c:([^(]+)\\(([0-9.]+)\\)", rank))[[1]]
67 if (length(match) == 3) {
68 result$class = match[2]
69 result$class_conf = as.numeric(match[3])
70 }
71 } else if (grepl("^o:", rank)) {
72 # Order
73 match = regmatches(rank, regexec("o:([^(]+)\\(([0-9.]+)\\)", rank))[[1]]
74 if (length(match) == 3) {
75 result$order = match[2]
76 result$order_conf = as.numeric(match[3])
77 }
78 } else if (grepl("^f:", rank)) {
79 # Family
80 match = regmatches(rank, regexec("f:([^(]+)\\(([0-9.]+)\\)", rank))[[1]]
81 if (length(match) == 3) {
82 result$family = match[2]
83 result$family_conf = as.numeric(match[3])
84 }
85 } else if (grepl("^g:", rank)) {
86 # Genus
87 match = regmatches(rank, regexec("g:([^(]+)\\(([0-9.]+)\\)", rank))[[1]]
88 if (length(match) == 3) {
89 result$genus = match[2]
90 result$genus_conf = as.numeric(match[3])
91 }
92 } else if (grepl("^s:", rank)) {
93 # Species
94 match = regmatches(rank, regexec("s:([^(]+)\\(([0-9.]+)\\)", rank))[[1]]
95 if (length(match) == 3) {
96 result$species = match[2]
97 result$species_conf = as.numeric(match[3])
98 }
99 }
100 }
101
102 return(result)
103}
104
105# read ASV table
106table = read.table(ASV_table, sep = "\t", check.names = F, header = T, row.names = 1)
107
108# read SINTAX taxonomy table (vsearch --sintax output format)
109# Format: ASV_ID \t taxonomy_string \t strand \t other_columns
110tax_raw = read.table(taxtab, sep = "\t", check.names = F, header = F,
111 stringsAsFactors = F, quote = "", comment.char = "", fill = TRUE)
112
113# Take first two columns only (ASV_ID and taxonomy)
114tax_raw = tax_raw[, 1:2]
115colnames(tax_raw) = c("ASV", "taxonomy")
116rownames(tax_raw) = tax_raw$ASV
117
118cat("\n Input =", nrow(tax_raw), "features.\n")
119
120# Parse SINTAX taxonomy strings
121tax_list = lapply(tax_raw$taxonomy, parse_sintax)
122tax = do.call(rbind, lapply(tax_list, as.data.frame))
123rownames(tax) = tax_raw$ASV
124
125# taxon list
126taxon_list = strsplit(target, ", ")[[1]]
127
128### extract only target-taxon ASVs from the 'raw' SINTAX results
129tax_filtered = tax %>%
130 filter(.data[[tax_level]] %in% taxon_list)
131
132cat("\n Found", nrow(tax_filtered), "ASVs matching", target, "at", tax_level, "level.\n")
133
134### change all tax ranks to "unclassified_*" when
135# the confidence values is less than the specified threshold
136# kingdom
137tax_filtered = tax_filtered %>%
138 mutate(kingdom = ifelse(kingdom_conf < threshold | is.na(kingdom),
139 "unclassified_root", as.character(kingdom)))
140
141# phylum
142tax_filtered = tax_filtered %>%
143 mutate(phylum = ifelse(phylum_conf < threshold | is.na(phylum),
144 paste0("unclassified_", kingdom), as.character(phylum)))
145tax_filtered$phylum = stringr::str_replace(tax_filtered$phylum, "unclassified_unclassified_",
146 "unclassified_")
147
148# class
149tax_filtered = tax_filtered %>%
150 mutate(class = ifelse(class_conf < threshold | is.na(class),
151 paste0("unclassified_", phylum), as.character(class)))
152tax_filtered$class = stringr::str_replace(tax_filtered$class, "unclassified_unclassified_",
153 "unclassified_")
154
155# order
156tax_filtered = tax_filtered %>%
157 mutate(order = ifelse(order_conf < threshold | is.na(order),
158 paste0("unclassified_", class), as.character(order)))
159tax_filtered$order = stringr::str_replace(tax_filtered$order, "unclassified_unclassified_",
160 "unclassified_")
161
162# family
163tax_filtered = tax_filtered %>%
164 mutate(family = ifelse(family_conf < threshold | is.na(family),
165 paste0("unclassified_", order), as.character(family)))
166tax_filtered$family = stringr::str_replace(tax_filtered$family, "unclassified_unclassified_",
167 "unclassified_")
168
169# genus
170tax_filtered = tax_filtered %>%
171 mutate(genus = ifelse(genus_conf < threshold | is.na(genus),
172 paste0("unclassified_", family), as.character(genus)))
173tax_filtered$genus = stringr::str_replace(tax_filtered$genus, "unclassified_unclassified_",
174 "unclassified_")
175
176# species to genus_sp when the confidence values is < species_threshold
177tax_filtered = tax_filtered %>%
178 mutate(species = ifelse(species_conf < species_threshold | is.na(species),
179 paste0(genus, "_sp"), as.character(species)))
180
181### exclude off-target taxa at any taxonomic level
182if (length(off_targets) > 0 && !all(is.na(off_targets)) && off_targets[1] != "") {
183 # Count ASVs before exclusion
184 n_before_exclusion = nrow(tax_filtered)
185
186 # Exclude ASVs where any taxonomic level matches off-targets
187 # Check all taxonomic ranks: kingdom, phylum, class, order, family, genus, species
188 tax_filtered = tax_filtered %>%
189 filter(!(kingdom %in% off_targets |
190 phylum %in% off_targets |
191 class %in% off_targets |
192 order %in% off_targets |
193 family %in% off_targets |
194 genus %in% off_targets |
195 species %in% off_targets))
196
197 n_after_exclusion = nrow(tax_filtered)
198 n_excluded = n_before_exclusion - n_after_exclusion
199
200 cat("\n Excluded", n_excluded, "ASVs matching off-target taxa at any taxonomic level:",
201 paste(off_targets, collapse = ", "), "\n")
202 cat(" Remaining ASVs after exclusion:", n_after_exclusion, "\n")
203}
204
205### count occurrences of each taxon in df (SINTAX results)
206count_taxa = function(df, taxa) {
207 sapply(taxa, function(taxon) sum(apply(df, 1, function(row) any(row == taxon))))
208}
209taxon_counts = count_taxa(tax_filtered, taxon_list)
210
211# Check the counts
212if (all(taxon_counts == 0)) {
213 print("ERROR: None of the specified taxa are present in the SINTAX results.")
214} else {
215 if (any(taxon_counts == 0)) {
216 warning("One or more of the specified taxa are not present in the SINTAX results.")
217 }
218 cat("\n Taxon counts:\n")
219 print(taxon_counts)
220}
221
222### extract only target-taxon ASVs from the 'threshold filtered' SINTAX results
223tax_filtered_thresh = tax_filtered %>%
224 filter(.data[[tax_level]] %in% taxon_list)
225
226# Remove confidence columns for output
227tax_filtered_output = tax_filtered_thresh %>%
228 select(kingdom, phylum, class, order, family, genus, species)
229
230# write filtered SINTAX taxonomy table
231tax_filtered_output = cbind(ASV = rownames(tax_filtered_output), tax_filtered_output)
232write.table(tax_filtered_output,
233 file = "taxonomy.sintax.filt.txt",
234 quote = F,
235 row.names = F,
236 sep = "\t")
237
238### filter the ASV table to match ASVs that were kept in the tax_filtered table
239table_filt = table[rownames(table) %in% rownames(tax_filtered_thresh), ]
240
241# write filtered table
242table_filt = cbind(ASV = rownames(table_filt), table_filt)
243write.table(table_filt,
244 file = paste0(sub("\\.[^.]*$", "_tax_filt.txt", ASV_table)),
245 quote = F,
246 row.names = F,
247 sep = "\t")
248
249# filter ASV_fasta
250fasta = readDNAStringSet(ASV_fasta)
251fasta.tax_filt = fasta[names(fasta) %in% rownames(table_filt)]
252
253# write filtered ASV_fasta
254writeXStringSet(fasta.tax_filt,
255 paste0(sub("\\.[^.]*$", "_tax_filt.fasta", ASV_fasta)),
256 width = max(width(fasta.tax_filt)))
Output files where off-target taxa have been removed:
taxonomy.sintax.filt.txt: filtered SINTAX taxonomy tableASVs_table_collapsed_tax_filt.txt: taxonomy filtered ASV tableASVs_collapsed_tax_filt.fasta: taxonomy filtered ASVs fasta file
Example of the filtered SINTAX taxonomy table:
ASV |
kingdom |
phylum |
class |
order |
family |
genus |
species |
|---|---|---|---|---|---|---|---|
46a9fb279afe3d45b304409d09d63e9181f94096 |
Metazoa |
Arthropoda |
Chilopoda |
Lithobiomorpha |
Lithobiidae |
Lithobius |
Lithobius_curtipes |
37838deee5cc9233c9ac7c74f01d8c06a7912bb1 |
Metazoa |
Arthropoda |
Arachnida |
Sarcoptiformes |
Liacaridae |
Adoristes |
Adoristes_ovatus |
c787bfb1fb95d92352fe1579dcc1a8f67c0d5ebe |
Metazoa |
Annelida |
Clitellata |
Enchytraeida |
Enchytraeidae |
Fridericia_segmented_worms |
Fridericia_eiseni |
e837216e5c192a25cf3808ba39f29c34b3fe00e9 |
Metazoa |
Arthropoda |
unclassified_Arthropoda |
unclassified_Arthropoda |
unclassified_Arthropoda |
unclassified_Arthropoda |
unclassified_Arthropoda_sp |
Note that the taxonomix ranks with lower bootstrap values than ``cutoff`` value have been changed to “unclassified_”.*
Additional processes via QuickTools
The following are additional steps that can be performed, with removing putative NUMTs as the most important one for COI data.
Remove NUMTs
When working with processing protein-coding markers (such as COI), the amplified sequences of nuclear mitochondrial pseudogenes (NUMTs) may inflate the richness estimates and thus introduce biases in biodiversity research using metabarcoding. Therefore, it is important to remove these sequences from the data set.
In PipeCraft2, there are tools such as metaMATE and ORFfinder that can be used to remove NUMTs from the data set.
Here, we use ORFfinder.
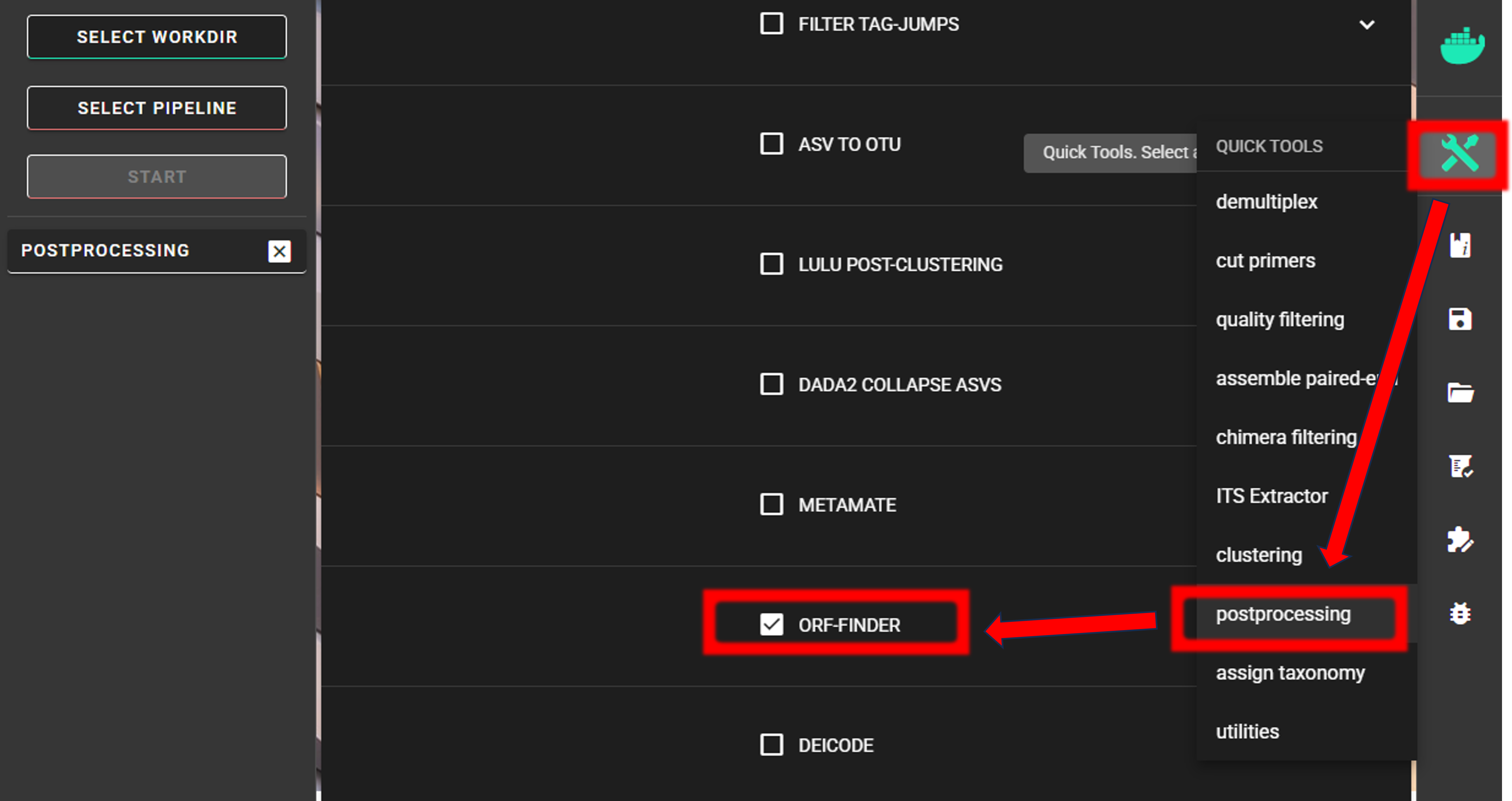
Here, input data is only fasta file. We are selecting out filtered fasta file ASVs_collapsed_tax_filt.fasta.
As we are interesed in “The Invertebrate Mitochondrial Code”, we as specifying 5 in the genetic code setting
(in PipeCraft, click on the genetic code setting to see the available genetic codes).
The min length and max length settings were already set in the CURATE ASV TABLE step, so here, those setting to
not have an effect unless we narrow down the accepdable length range.
Double-check the selected working directory (the outputs will be written there) by holding
the mouse cursor over the SELECT WORKDIR button [re-select if needed].
Press “START” to start the ORFfinder process.
Output files: |
|
|---|---|
*_ORFs.fasta |
fasta file of filtered ASVs |
*_ORFs.list.txt |
list of of filtered ASVs |
*_notORFs.fasta |
fasta file of ASVs that did not pass ORF-finder |
*_notORFs.list.txt |
list of ASVs that did not pass ORF-finder |
This process filters only the fasta file and as a main output it creates a a list of ASVs that passed and did not pass the genetic code translation.
In this example dataset, ORFfinder identidied 1 ASV that did not pass the genetic code translation. Let’s discard this ASV from the dataset.
Below, you can find a R script to clean also taxonomy and ASV tables.
1#!/usr/bin/env Rscript
2### Filter dataset based on ORF-finder results to exclude putative NUMTs
3
4# Specify input files
5discard_file = "ASVs_collapsed_tax_filt.notORFs.list.txt"
6fasta_file = "ASVs_collapsed_tax_filt.fasta"
7ASV_table_file = "ASVs_table_collapsed_tax_filt.txt"
8taxonomy_file = "taxonomy.sintax.filt.txt"
9#--------------------------------------#
10library(seqinr)
11
12# Read the list of ASVs that should be discarded based on ORFfinder output
13discard = read.table(discard_file, header = FALSE)
14# Read the fasta file
15fasta = read.fasta(fasta_file)
16n_input = length(fasta)
17# Read the ASV table
18ASV_table = read.table(ASV_table_file, header = TRUE)
19# Read the taxonomy file
20taxonomy = read.table(taxonomy_file, header = TRUE)
21### Remove ASVs from fasta file
22fasta = fasta[!names(fasta) %in% discard$V1]
23
24# Remove ASVs from ASV table
25ASV_table = ASV_table[!ASV_table$ASV %in% discard$V1, ]
26
27# Remove ASVs from taxonomy file
28taxonomy = taxonomy[!taxonomy$ASV %in% discard$V1, ]
29
30# Summary
31cat("Number of input ASVs:", n_input, "\n")
32cat("Number of ASVs discarded:", nrow(discard), "\n")
33cat("Number of ASVs left:", length(fasta), "\n")
34
35# Create output folder
36outdir <- "ORF_filtered"
37if (!dir.exists(outdir)) {
38 dir.create(outdir)
39}
40
41# Output names: input basename with "_ORFs" before extension
42add_ORFs <- function(f) {
43 paste0(tools::file_path_sans_ext(basename(f)), "_ORFs.", tools::file_ext(f))
44}
45
46# Write outputs
47write.fasta(fasta, names(fasta), nbchar = 999,
48 file.path(outdir, add_ORFs(fasta_file)))
49write.table(ASV_table, file.path(outdir, add_ORFs(ASV_table_file)),
50 sep = "\t", quote = FALSE, row.names = FALSE)
51write.table(taxonomy, file.path(outdir, add_ORFs(taxonomy_file)),
52 sep = "\t", quote = FALSE, row.names = FALSE)
Output files
ORF_filtereddirectory:ASVs_collapsed_tax_filt_ORFs.fasta: fasta file of filtered ASVsASVs_table_collapsed_tax_filt_ORFs.txt: filtered ASV tabletaxonomy.sintax.filt_ORFs.txt: filtered taxonomy table
Proceed with any relevant statistical analyses using these filtered files if you are interesed in ASV-level analyses, or proceed with clustering ASVs into OTUs (see below).
Cluster ASVs into OTUs
If the aim is not to do the haplotype-level analyses, then ASVs can be clustered into OTUs (PipeCraft uses vsearch for this). The ASVs approach may not accurately reflect species composition in the community of as COI gene has highly variable levels of intraspecific polymorphism. Thus, one species may be represented by many ASVs, whereas other species may be represented by very few ASVs.
Here, we are clustering ASVs to OTUs (using vsearch) via QuickTools panel (on the right ribbon).
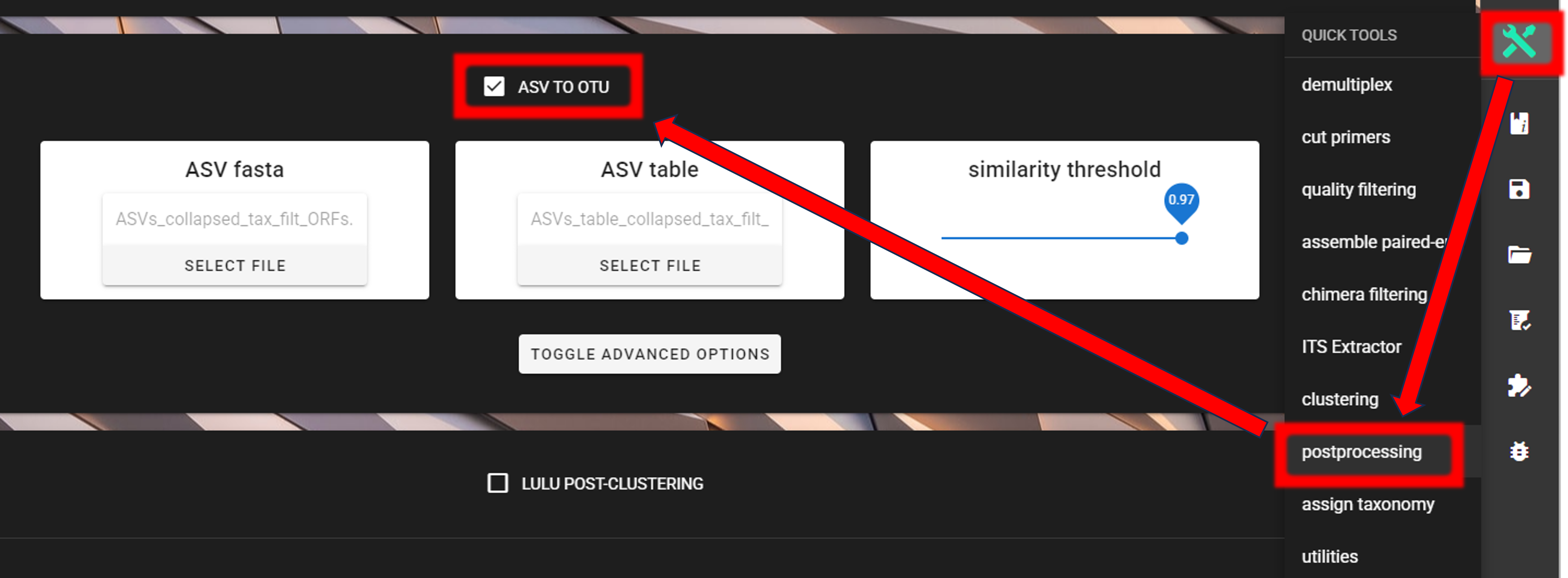
Input data are files in the ORF_filtered directory:
ASVs_collapsed_tax_filt_ORFs.fastaASVs_table_collapsed_tax_filt_ORFs.txt
As a similarity threshold we are using the commonly used 97% (0.97, default setting).
Note
2nd column of the input ASV table must be ‘Sequences’ (1st column is ASV IDs; this is the default PipeCraft2 output table, so you don’t need to worry about this if you have followed this pipeline). For clustering, the ASV size annotation is obtained from the ASV table.
If the ASV table does not contain ‘Sequence’ column, then add those with QuickTools -> Utilities -> Add sequences to table
see here.
Specify ORF_filtered directory as a working directory via SELECT WORKDIR button and press “START” to start the process.
Outputs in |
|
|---|---|
OTUs.fasta |
FASTA formated representative OTU sequences |
OTU_table.txt |
OTU table (tab delimited file) |
OTUs.uc |
uclust-like formatted clustering results |
Herein, clustering formed 17 OTUs from 19 ASVs (with 97% similarity threshold).
As we performed taxonomy annotation to ASVs, we can now match the taxonomy to the OTUs.
1#!/usr/bin/env Rscript
2### Get taxonomy for OTUs based on ASVs taxonomy
3
4# Specify input files
5# Working directory is "ASVs2OTUs_out"
6ASV_taxonomy_file = "../taxonomy.sintax.filt_ORFs.txt"
7OTU_fasta_file = "OTUs.fasta"
8#--------------------------------------#
9library(seqinr)
10
11# Read the ASV taxonomy file
12ASV_taxonomy = read.table(ASV_taxonomy_file, header = TRUE)
13# Read the OTU fasta file
14OTU_fasta = read.fasta(OTU_fasta_file)
15# Get the taxonomy for each OTU
16OTU_taxonomy = ASV_taxonomy[ASV_taxonomy$ASV %in% names(OTU_fasta), ]
17
18# Write the OTU taxonomy file
19write.table(OTU_taxonomy, file = "taxonomy.OTUs.txt", sep = "\t",
20 quote = FALSE, row.names = FALSE, col.names = TRUE)
OTU taxonomy file
taxonomy.OTUs.txt
When now comparing the ASVs and OTUs taxonomy tables, we can see that the ASVs taxonomy table had 2 ASVs assigned to Euzetes globulus species, and 2 ASVs assigned to Holoparasitus calcaratus species, but those are now merged respectively into 1 OTU for each of these species (with 97% similarity threshold).
However, in the OTUs taxonomy table, we see that Lithobius curtipes and Adoristes ovatus are represented by 2 OTUs, respectively. Certanly, the barcoding gaps may vary between different specie, thus resulting in different OTUs for the same species when using single sequence similarity threshold. But let’s check if the post-clustering process will help to merge these OTUs with same species names (see below).
LULU post-clustering
Additionally, we can perform LULU post-clustering to merge co-occurring ‘daughter’ OTUs.
LULU description from the LULU repository: the purpose of LULU is to reduce the number of erroneous OTUs in OTU tables to achieve more realistic biodiversity metrics. By evaluating the co-occurence patterns of OTUs among samples LULU identifies OTUs that consistently satisfy some user selected criteria for being errors of more abundant OTUs and merges these. It has been shown that curation with LULU consistently result in more realistic diversity metrics.
Here, we are performing LULU post-clustering via QuickTools panel (on the right ribbon).
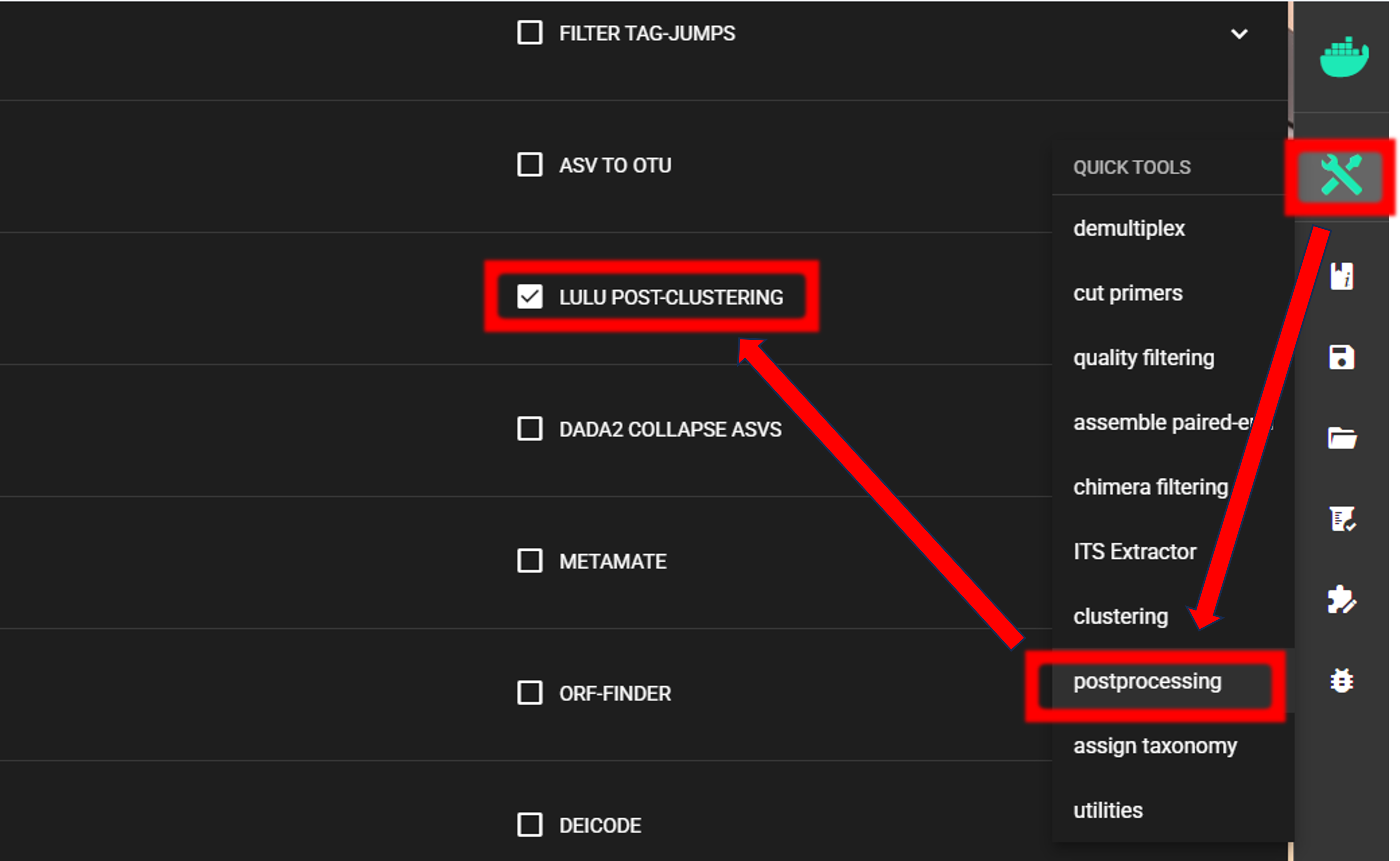
The input data are OTU_table.txt and OTUs.fasta files in the ASVs2OTUs_out directory.
Here, we are using the default settings (which are suitable for most cases),
but feel free to experiment with various settings to see the effect on the results.

To START
To START, specify working directory under SELECT WORKDIR (outputs will be written here),
but the following requests about Sequence files extension and Sequencing read types do not matter here, just click ‘Confirm’.
Outputs in |
|
|---|---|
OTU_table.lulu.txt |
curated table in tab delimited txt format |
OTUs.lulu.fasta |
fasta file for the molecular units (OTUs or ASVs) in the curated table |
match_list.lulu |
match list file that was used by LULU to merge ‘daughter’ molecular units |
discarded_units.lulu
|
molecular units (OTUs or ASVs) that were merged with other units based on
specified thresholds
|
The input for LULU post-clustering had 17 OTUs, with double representation of Lithobius_curtipes and Adoristes_ovatus OTUs:
OTU |
sample1_COI |
sample2_COI |
sample3_COI |
|---|---|---|---|
Adoristes_ovatus
Adoristes_ovatus
Entomobrya_sp
Euzetes_globulus
Fridericia_eiseni
Holoparasitus_calcaratus
Lithobius_curtipes
Lithobius_curtipes
Orchesella_flavescens
Pergalumna_nervosa
Tomocerus_sp
Trachytes_aegrota
unclassified_Arthropoda_sp
unclassified_Arthropoda_sp
unclassified_Arthropoda_sp
unclassified_Arthropoda_sp
unclassified_Metazoa_sp
|
0
0
0
0
215
0
15
583
0
0
0
0
0
0
0
0
0
|
342
0
0
179
0
38
0
0
0
45
0
7
75
81
0
0
0
|
0
3
25
0
0
7
0
0
10
0
35
0
0
0
39
682
2
|
After LULU post-clustering, LULU merged 2 Lithobius_curtipes OTUs into a single Lithobius_curtipes OTU. But Adoristes ovatus remains represented as 2 OTUs.
OTU |
sample1_COI |
sample2_COI |
sample3_COI |
|---|---|---|---|
Adoristes_ovatus
Adoristes_ovatus
Entomobrya_sp
Euzetes_globulus
Fridericia_eiseni
Holoparasitus_calcaratus
Lithobius_curtipes
Orchesella_flavescens
Pergalumna_nervosa
Tomocerus_sp
Trachytes_aegrota
unclassified_Arthropoda_sp
unclassified_Arthropoda_sp
unclassified_Arthropoda_sp
unclassified_Arthropoda_sp
unclassified_Metazoa_sp
|
0
0
0
0
215
0
598
0
0
0
0
0
0
0
0
0
|
342
0
0
179
0
38
0
0
45
0
7
75
81
0
0
0
|
0
3
25
0
0
7
0
10
0
35
0
0
0
39
682
2
|
In addition to sequence similarity, LULU post-clustering merges OTUs based on co-occurrence patterns, and as Adoristes ovatus OTUs are not co-occurring in the same samples, LULU did not merge them. On the other hand, Lithobius curtipes OTUs were both in sample1_COI, thus were merged into single Lithobius curtipes OTU by summing up the abundances of the two OTUs.
The last step
The last step here is to get matching taxonomy table for the LULU-curated OTUs.
1#!/usr/bin/env Rscript
2### Get final OTUs taxonomy
3
4# Specify input files
5OTUs_taxonomy_file = "taxonomy.OTUs.txt" # OTUs taxonomy table
6OTU_fasta_file = "OTUs.lulu.fasta" # LULU-curated OTUs fasta file
7#--------------------------------------#
8library(seqinr)
9
10# Read the ASV taxonomy file
11OTUs_taxonomy = read.table(OTUs_taxonomy_file, header = TRUE)
12# Read the OTU fasta file
13OTU_fasta = read.fasta(OTU_fasta_file)
14# Get the taxonomy for each OTU
15OTU_taxonomy = OTUs_taxonomy[OTUs_taxonomy$ASV %in% names(OTU_fasta), ]
16
17# Write the OTU taxonomy file
18write.table(OTU_taxonomy, file = "taxonomy.OTUs.lulu.txt",
19 sep = "\t", quote = FALSE, row.names = FALSE)
Final curated OTU files
Now, final curated files are:
taxonomy.OTUs.lulu.txtOTUs.lulu.fastaOTU_table.lulu.txt
Proceed with any relevant statistical analyses using the curated files.